馬偕兒童醫院,過敏免疫風濕科 鄭勇鋒/徐世達醫師
背景/目的
局限性硬皮病與系統性硬皮症皆以皮膚纖維化和/或其他器官系統受累為特徵。本文回顧馬偕兒童醫院兒童局限性硬皮病與系統性硬皮症患者的臨床表現、皮膚外的併發症及傳統治療選擇。
方法
自1993年3月至2023年10月,於台北馬偕紀念醫院兒科部共收錄42位診斷為系統性硬皮症(systemic sclerosis;SSc)或局限性硬皮病(localized scleroderma;LSc)的病童。診斷依據美國風濕病醫學會診斷標準及臨床皮膚硬化表現分類。收集資料包括性別、實驗室數據、家族史、外傷史、治療方式及預後。
結果
共有3位患者(0.71%)罹患SSc,39位患者(92.86%)為LSc,其中女生31位(73.8%)、男生11位(26.2%)。抗核抗體在22位患者(52.4%)呈陽性;抗Scl-70抗體在3位患者(7.1%)呈陽性。一名男孩合併軍刀型硬皮病(en coup de sabre)及後顱窩腫瘤。三位SSc患者出現雷諾氏現象。最常見的皮膚外併發症為肌肉骨骼系統受累。37位患者(90.4%)接受D-青黴胺治療;大多數病例使用甲氨蝶呤;32位患者接受口服類固醇治療。一名LSc女孩在接受D-青黴胺治療後出現蛋白尿。所有局限性患者最終均記錄到皮膚病灶軟化。
結論
局限性硬皮病患者死亡率低,但罹病率顯著。損害不僅限於皮膚,許多患者有皮膚外系統受累。然而,LSc患者通常不會進展為系統性硬皮症。
關鍵詞: 兒童硬皮病、系統性硬皮症、線狀硬皮病、斑狀硬皮病、軍刀型硬皮病、D-青黴胺
引言
皮膚纖維化相關的兩種不同疾病實體為局限性硬皮病(LSc)與系統性硬皮症(SSc)。這兩種疾病可能屬於同一疾病譜系,但臨床病程卻有所不同。SSc傾向於在遠端肢體出現彌漫性皮膚纖維化,並伴隨臟器纖維化;LSc則多表現為線狀或局限性皮膚病灶,並可能合併皮膚外病灶,其中最常見的是關節炎。SSc在成人的盛行率高於兒科患者。兒科患者的死亡率低於成人患者。雖然兒童LSc的死亡率低,但其罹病率顯著。有極少數LSc患者會進展為SSc。本研究旨在呈現42例兒科硬皮病患者的臨床特徵與預後。
方法
我們回顧了1993年3月至2023年10月間,在台北馬偕紀念醫院兒科部診斷為SSc或LSc病童的病歷。本研究無排除標準,共納入42位患者,其中3位為SSc,39位為LSc。SSc和LSc的診斷係依據美國風濕病學會(ACR)標準。皮膚受累依據LeRoy分類分為侷限型(手、前臂、臉或足)或瀰漫型(軀幹及肢端)。[1] 收集資料包括性別、臨床表現、實驗室數據、家族史、外傷史、治療方式及預後。抗核抗體(ANA)、抗Scl-70抗體及抗雙股DNA抗體(anti-dsDNA)以雙向免疫擴散法檢測。類風濕因子(RF)以散射光度法測定。疾病病程為透過病歷回顧或患者及家屬電話訪談進行評估。
結果
共有3位患者(0.71%)罹患SSc,39位患者(92.86%)為LSc,其中女生31位(73.8%)、男生11位(26.2%)(表1)。部分患者曾於其他醫院接受治療,部分則未曾治療。一名男孩合併軍刀型硬皮病(en coup de sabre)及後顱窩腫瘤。三位SSc患者(病例1、2、12)出現雷諾氏現象。最常見的皮膚外併發症為肌肉骨骼系統受累。一名女孩(病例32)患有軍刀型硬皮病並有口腔內受累。一名男孩(病例40)左腿泛硬皮病導致患肢比右腿短3公分。一名SSc女孩出現肺纖維化。病例5與病例7在外傷後皮膚病灶惡化。一名患者出現單側廣泛性斑狀硬皮病。[2] 抗核抗體在22位患者(52.4%)呈陽性;抗Scl-70抗體在3位患者(7.1%)呈陽性。一名LSc患者(病例7)有姑媽罹患類風濕性關節炎;病例39的祖母罹患SSc。32位患者(90.4%)接受D-青黴胺治療;大多數病例使用甲氨蝶呤;32位患者接受口服類固醇治療。一名LSc女孩在D-青黴胺治療後出現蛋白尿,因而停藥D-青黴胺。所有局限性患者最終均記錄到皮膚病灶軟化。
表 1:42例病童資料彙整表

PS: Prednisolone(潑尼松龍)、D-PC:D-青黴胺、MTX:甲氨蝶呤
ANA: 抗核抗體,正常範圍(<1:80) RF: 類風濕因子,正常範圍(<20 IU/mL) RA: 類風濕性關節炎 (S): 顆粒型螢光圖案 (H): 均質型螢光圖案 (N): 核仁型螢光圖案 (S/H): 顆粒至均質型螢光圖案
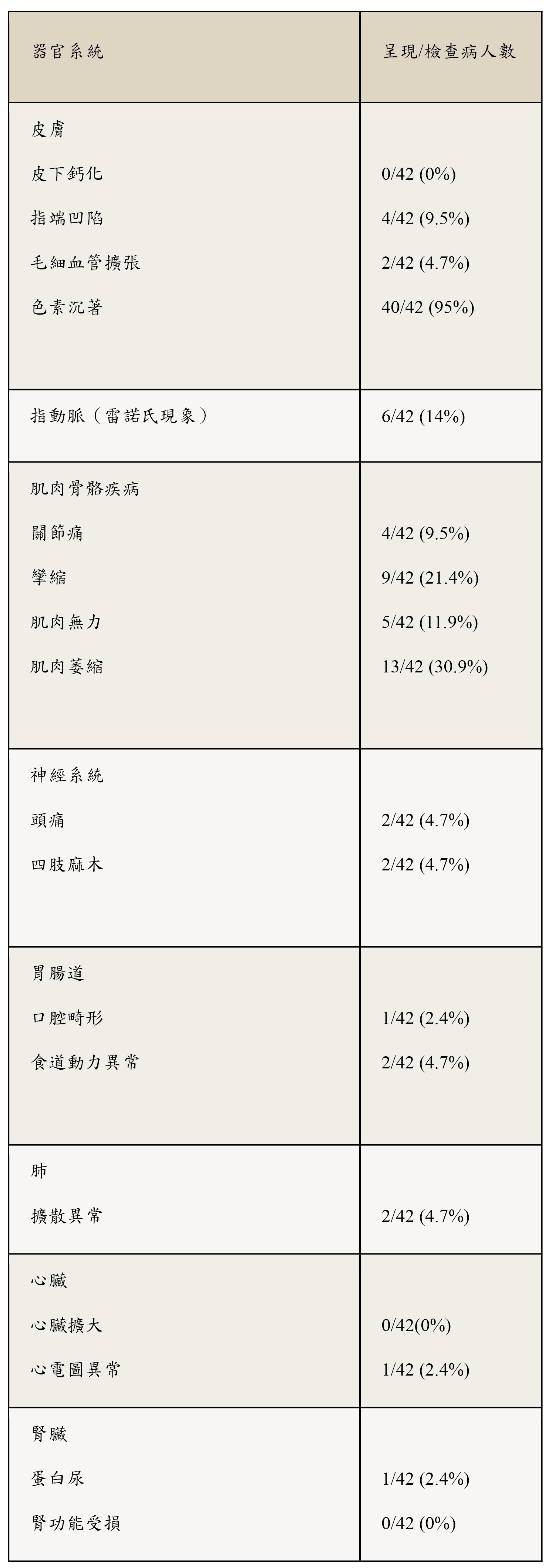
器官系統受侵犯情形列於表2,其中最常見的皮膚外表現為肌肉骨骼系統受累。
表 2:
討論
美國兒科硬皮病的年度盛行率估計約為每一萬名兒童中有 3.2 至 3.6 例。[3] 在台灣,系統性硬皮症(SSc)的年度發生率為每百萬人口 10.9 例(男性 4.7 例,女性 17.4 例)。2002 至 2007 年間,平均盛行率為每百萬人口 56.3 例。[4] 兒童 SSc 極為罕見。
已知遺傳因素與環境因素共同影響局限性硬皮病(LSc)的病理生理。在具遺傳易感性的兒童中,環境因素可能觸發疾病的發生與進展。來自 SSc 患者皮膚切片與外周血的研究顯示,免疫機制、細胞激素功能、免疫細胞功能、血管與纖維母細胞活動均有異常。纖維化與抗纖維化途徑的失調導致纖維母細胞持續活化與上皮-間質轉化,造成過度的細胞外基質合成與膠原沉積。纖維母細胞對轉化生長因子 β1(TGF-β1)的反應減弱,並更多分化為肌成纖維細胞。TGF-β 超家族的關鍵調節因子(如 SOSTDC1 與 FRAS1)也被發現失調。重要的是,分泌組(secretome)的調控能改變纖維母細胞表型,顯示分泌組調控可能成為潛在的治療靶點。
T 輔助細胞(Th1)的過度活化、腫瘤壞死因子α的過度產生與反應,以及 B 細胞、T 細胞與調節性 T 細胞的調控不良,導致皮膚組織的炎症。CCL18 已被證實是疾病活動的特異性預測因子,其表現優於 CXCL9 或 CXCL10。研究顯示,在炎症病灶邊緣,CCL18 基因表達顯著高於硬化中心、未受影響的皮膚或健康對照。[5]
SSc 與 LSc 患者的血清與內皮細胞均顯示內皮細胞黏附分子過度表達。血管細胞黏附分子 1(VCAM-1)與 E-selectin 在 LSc 患者中顯著高於健康對照,並與硬皮病灶數量及受累範圍相關。[5]
LSc 與 SSc 的皮膚病灶在組織學與外觀上非常相似,雖然分布不同。它們均經歷初期炎症期、纖維化期與後期階段。初期病灶可能表現為紅斑或蠟樣硬化區周圍的藍色調。水腫或紅斑病灶之後可能出現硬化、低色素或高色素變化,以及皮膚萎縮。
在局限性硬皮病中,皮膚病灶可能表現為斑狀硬皮病(橢圓或圓形病灶)或線狀硬皮病(線狀病灶)。LSc 可依皮膚病灶特徵分類為斑狀型、廣泛型與線狀型。若病灶累及兩個或以上解剖區域,則為廣泛型。[5] LSc 病灶大小可從數公分至整個肢體長度不等,深度可累及皮膚與組織。線狀 LSc 傾向侵犯更深層組織,如皮下組織、筋膜、肌肉,甚至骨骼與關節。此外,關節炎與關節痛可能發生在遠離皮膚病灶的關節。部分 LSc 患者病灶僅限於皮膚,部分則合併皮膚外病灶。
在系統性硬皮症中,疾病標誌為雙手手指皮膚增厚,延伸至掌指關節近端,並伴隨臟器纖維化。肺纖維化可能導致肺功能不良、肺動脈高壓與右心衰竭。SSc 患者更常合併自體免疫疾病,並可能在風濕免疫相關抗體檢測中呈陽性,如 ANA、RA 及其他 ENA。硬皮病相關抗體包括抗著絲點抗體與抗拓撲異構酶 I 抗體,後者為疾病嚴重程度的指標。這些患者可能出現雷諾氏現象與甲床毛細血管異常。[6] 肺纖維化是預後不良的指標,可能導致死亡,儘管兒科患者的 SSc 存活率優於成人。
在我們的經驗中,病例 42 主訴左頸與鎖骨上區皮膚病灶,但在門診檢查時,發現其左額有線狀病灶,患者與家屬均未察覺。病例 40 在就診前即出現萎縮與左腿比右腿短 3 公分。病例 32 有臉部線狀硬皮病(軍刀型硬皮病),並伴隨口腔病灶,包括牙齦萎縮與舌萎縮。(圖 1:(A)左側嘴唇與下巴的線狀硬皮病;(B)左側舌萎縮)一名 SSc 患者因治療依從性差而進展為肺纖維化。早期診斷與適當治療對於預防 LSc 患者殘疾至關重要。
圖 1. 左側嘴唇與下巴的線狀硬皮病
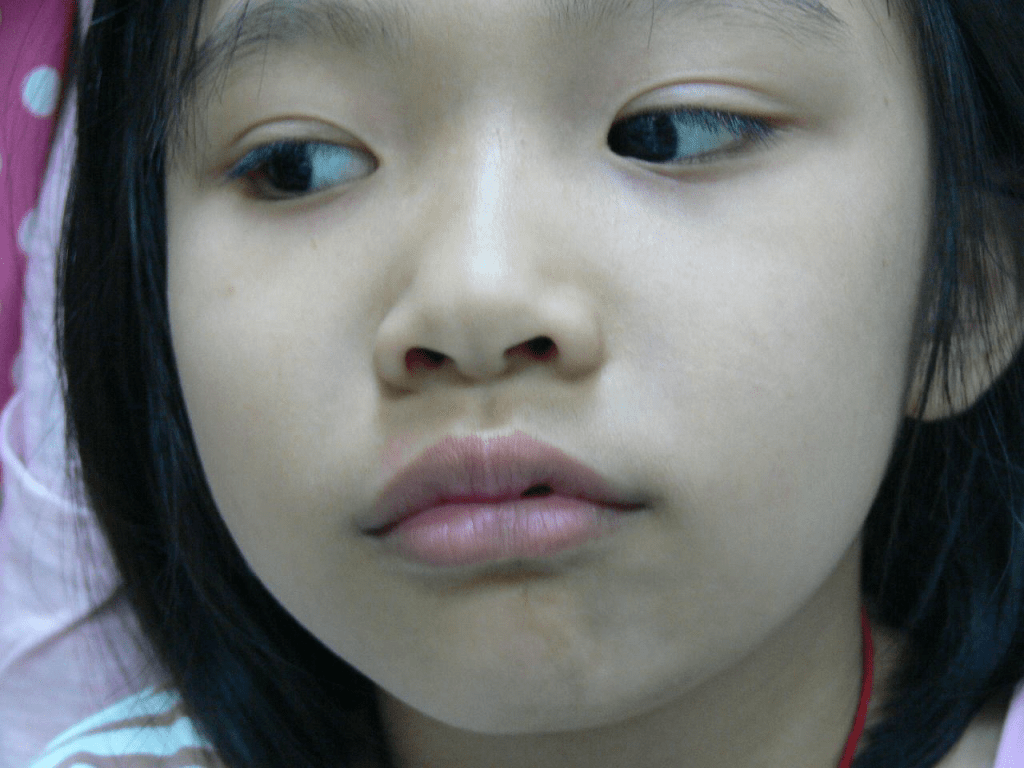
圖 2. 左側舌頭萎縮

目前並沒有特定的血液檢查或影像學檢查可以診斷硬皮病。自體免疫疾病的抗體檢測可用於鑑別診斷及判斷是否合併其他自體免疫疾病。對於軍刀型硬皮病(en coup de sabre)患者,可能需要進行 MRI 檢查以評估中樞神經系統受累,這些患者可能會有頭痛或癲癇發作。局限性硬皮病患者的診斷或治療並不需要皮膚切片。
適當的治療對於預防硬皮病患者的死亡與罹病率至關重要。臨床醫師應在疾病進展風險與藥物治療負擔之間取得平衡。(圖 2:局限性硬皮病患者的初始治療)[7] 在患者就診時,可能處於初期炎症期、纖維化期或疾病不活躍期。治療選擇可從局部外用藥物到全身性藥物不等。所有硬皮病或硬皮症患者皆應進行皮膚護理,包括保濕、防止外傷及皮膚按摩。對於僅有局限性斑狀硬皮病,且未累及皮膚深層、未跨越關節或合併關節炎者,可使用局部糖皮質激素或他克莫司治療,並可考慮合併光療。在兒科患者中,紫外線治療需謹慎評估其副作用風險。
系統性治療應盡早開始,以防止兒童殘疾。尤其是臉部與頭皮的線狀 LSc,存在深層組織受累及中樞神經系統併發症的風險。對於病灶持續進展、累及關節或深層組織、或廣泛性斑狀硬皮病患者,應考慮系統性藥物治療。口服藥物的一線治療可採用全身性類固醇合併治療 3–6 個月,若可能應逐漸減量停用,並持續使用甲氨蝶呤 12–24 個月,依緩解狀態調整。D-青黴胺可用於硬皮病患者,其可能機制為透過整合素訊號負向調控纖維母細胞膠原基因表達,以及在小鼠纖維化基因中的作用。[8] 其他藥物在少數病例報告中被證實對甲氨蝶呤無效患者有益,包括生物製劑與JAK抑制劑。[5]
部分患者因畸形或關節受限可能需要手術。早期成功治療可避免畸形。[5] 手術應延後至疾病活動減退且生長過程完成,否則可能導致癒合延遲或受損,並需再次修正手術。
有雷諾氏現象的患者應使用血管擴張劑,如鈣離子通道阻斷劑或 ACEI,以防止指尖壞死。兒童 LSc 的預後良好。在兒童 LSc 患者中,疾病活動可能在數年炎症期後自行停止,無論是否接受治療;但若治療延誤,損害可能不可逆,並影響生活品質。
總結而言,SSc 與 LSc 在兒童的病程與預後不同。LSc 並非僅限於皮膚的疾病,皮膚外病灶可能導致患者殘疾。應盡早開始適當治療,並提高臨床醫師對此疾病的警覺,即使硬皮病的發生率並不常見。
